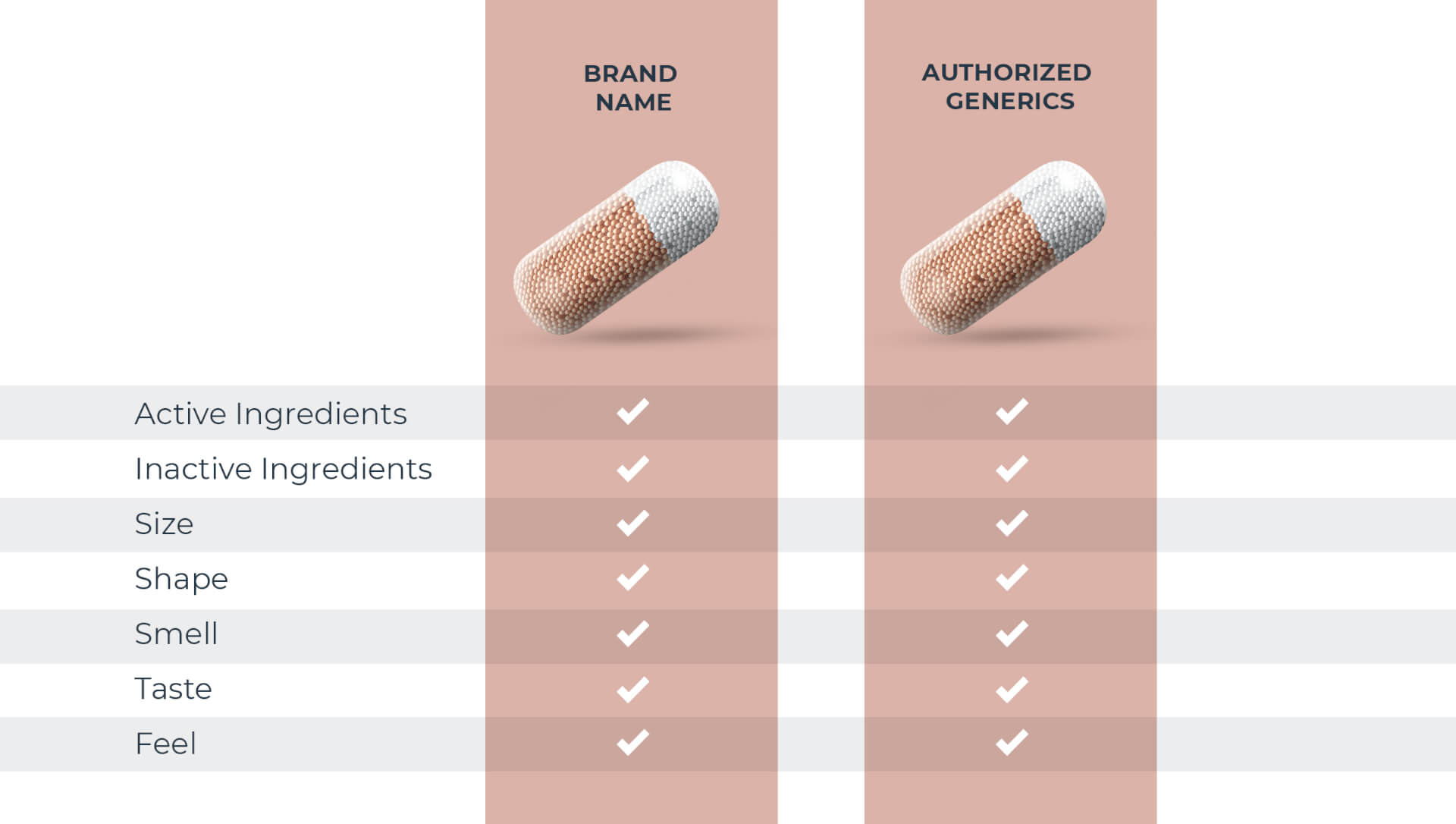
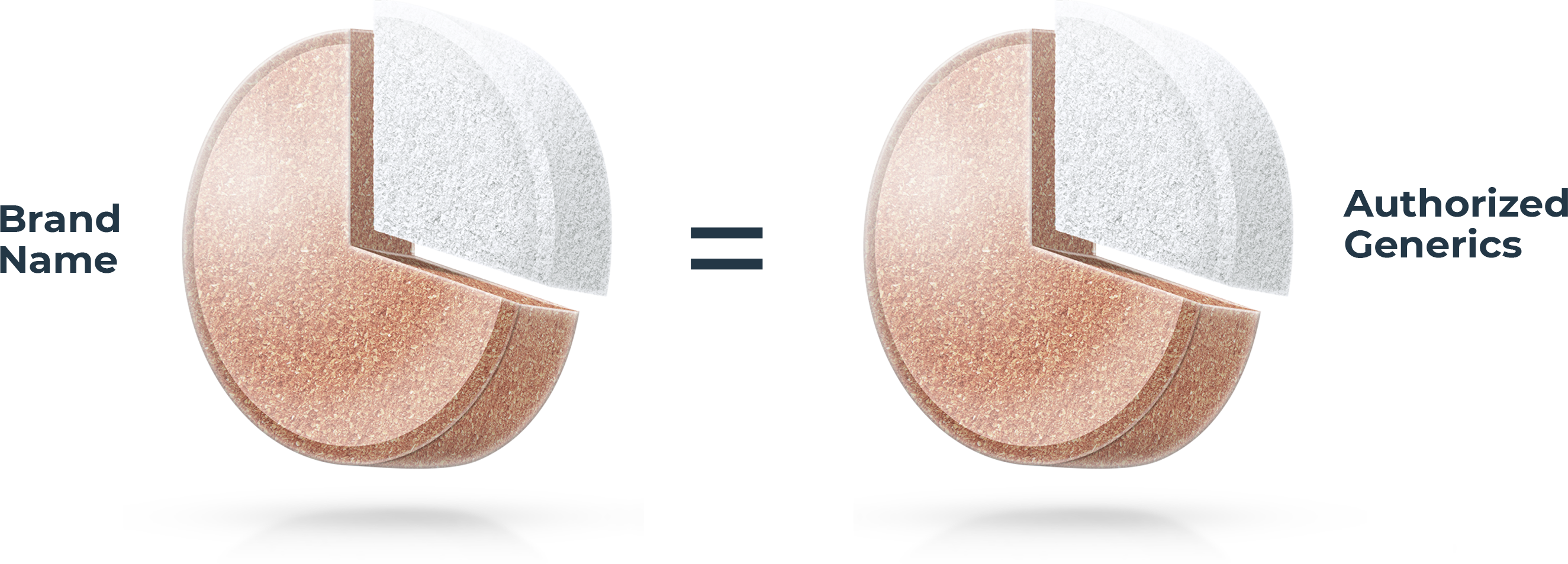Not all generic drugs have the same formula
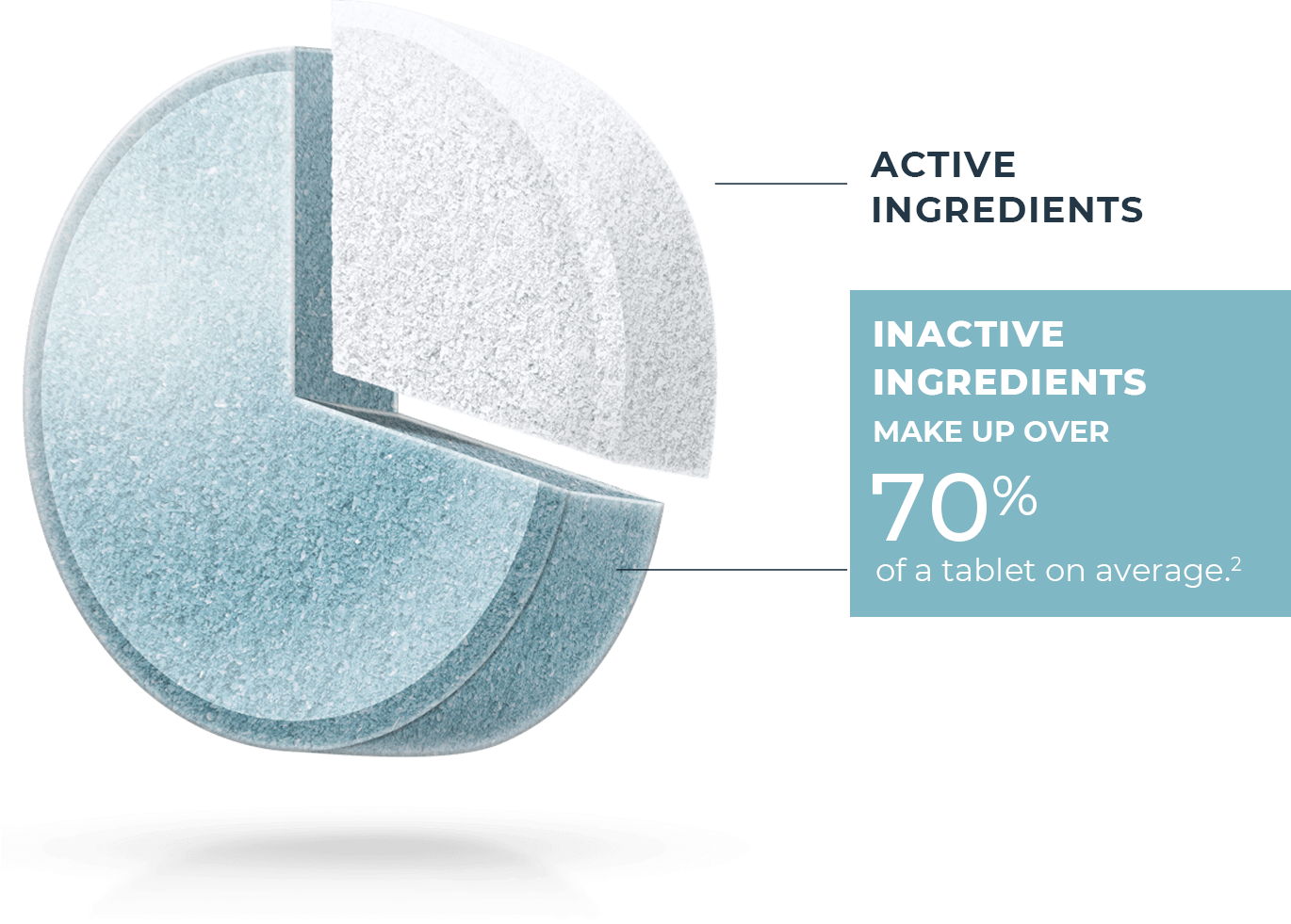
All generic drugs are FDA approved and are as safe and effective as the brand name. Most people know they also have the same active ingredients as the brand name, but many don’t realize generics are not required to have the same inactive ingredients1 as the brand. These make up over 70% of a tablet on average.